1 Регистрация
Для регистрации в системе перейдите по ссылке https://app.syntelly.com/ и нажмите кнопку «Зарегистрироваться».
На появившейся странице нажмите кнопку «Начать регистрацию»
Если ключа организации нет – то вам нужно будет пройти еще несколько шагов, заполнить данные анкеты и отправить заявку на тестовый доступ.
Введите адрес электронной почты, номер телефона и придумайте пароль для входа в Систему. Если у вашей организации уже есть оплаченный лицензионный доступ включите тумблер «Ключ продукта» и введите ключ, присланный вам ответственным лицом от вашей организации.
2 Вход в систему
Для входа в систему перейдите по ссылке https://app.syntelly.com/, введите адрес электронной почты, пароль и нажмите кнопку «Войти»:
После входа отображается раздел «Поиск»:
В меню слева можно выбрать раздел для работы, язык (русский или английский), открыть личный кабинет пользователя. Чтобы скрыть меню, нажмите кнопку.
В правом нижнем углу при открытии страницы возникают всплывающие подсказки по работе с Системой. Подсказка всплывает один раз в сутки. Закрыть подсказку можно, нажав на крестик. Полностью отключить подсказки можно в настройках профиля пользователя. Также в правом нижнем углу по клику открывается форма обратной связи для отправки письма в техническую поддержку компании Синтелли.
В правом нижнем углу при открытии страницы возникают всплывающие подсказки по работе с Системой. Подсказка всплывает один раз в сутки. Закрыть подсказку можно, нажав на крестик. Полностью отключить подсказки можно в настройках профиля пользователя. Также в правом нижнем углу по клику открывается форма обратной связи для отправки письма в техническую поддержку компании Синтелли.
3 Раздел «Поиск»
В этом разделе реализован доступ к основной базе данных, содержащей органические соединения, реакции, а также ссылки на публикации по химической тематике.
Чтобы открыть раздел, в панели слева нажмите «Поиск».
Чтобы открыть раздел, в панели слева нажмите «Поиск».
3.1 Поиск по структурам (молекулам)
Молекулы можно искать двумя способами:

- В молекулярном редакторе нарисовать структурную формулу молекулы. Для этого нажмите кнопку «Нарисовать». В открывшемся окне редактора нарисуйте молекулу и нажмите «Сохранить»:

После этого запустите поиск, нажав Enter или значок поиска.

Дополнительно можно установить следующие параметры поиска в панели «Фильтры» (панель автоматически отображается после первого поиска):
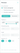
«Тип соответствия» — фильтр используется для нахождения структур по соответствию заданной молекуле:
«Точное совпадение» (найденная молекула должна полностью соответствовать заданной структуре),
«Подструктурный поиск» (проводится поиск молекул, содержащих нарисованный фрагмент),
«Похожие структуры» (выполняется поиск по молекулярному подобию);
«Подобие, %» — этот ползунок устанавливает пороги молекулярного подобия. Молекулярное подобие рассчитывается по методу Танимото на основе фингерпринтов ECFP. Перемещение ползунка изменяет числовое значение порога молекулярного подобия. Это значение определяет, насколько близко молекула должна соответствовать заданным критериям, чтобы считаться "похожей".
Для удаления всех настроенных параметров нажмите «Очистить все фильтры».
«Точное совпадение» (найденная молекула должна полностью соответствовать заданной структуре),
«Подструктурный поиск» (проводится поиск молекул, содержащих нарисованный фрагмент),
«Похожие структуры» (выполняется поиск по молекулярному подобию);
«Подобие, %» — этот ползунок устанавливает пороги молекулярного подобия. Молекулярное подобие рассчитывается по методу Танимото на основе фингерпринтов ECFP. Перемещение ползунка изменяет числовое значение порога молекулярного подобия. Это значение определяет, насколько близко молекула должна соответствовать заданным критериям, чтобы считаться "похожей".
Для удаления всех настроенных параметров нажмите «Очистить все фильтры».

- В поисковой строке можно ввести название молекулы в любом удобном формате (SMILES, синонимы, название по номенклатуре IUPAC, CAS-номер и кодовые обозначения из других баз данных) и нажать Enter или значок поиска.
- Чтобы очистить поисковую строку, нажмите на крестик в правой части поисковой строки
- При необходимости изменить молекулу нажмите кнопку
Подсказки:
3.1.1 Работа с найденными молекулами
Вы можете настроить масштаб карточек в отображаемом списке найденных молекул с помощью бегунка:
Для каждой карточки отображается:
Система позволяет:
Для выполнения нужного действия нажмите кнопку
в правом верхнем углу карточки и выберите соответствующую команду:
- ID молекулы в базе данных Синтелли;
- Структурная формула молекулы;
- Светофор – параметры безопасности молекулы с точки зрения токсичности («Tox»), физико-химических («Phys»), биологических («Bio») и экологических («Eco») свойств:
- Красный - молекула с высокими показателями опасности;
- Зеленый - молекула с низкими показателями опасности;
- Желтый - молекула со средними показателями опасности;
- «Литература» — ссылка на список документов, в которых встречается данное соединение;
- «Реакции» — ссылка на реакции, в которых участвует конкретное соединение.
Система позволяет:
- быстро скопировать SMILES;
- скопировать ссылку на карточку;
- открыть молекулу в молекулярном редакторе;
- скачать изображение структуры в формате .png;
- добавить молекулу в другой датасет.
Для выполнения нужного действия нажмите кнопку
в правом верхнем углу карточки и выберите соответствующую команду:
Чтобы посмотреть подробную информацию по молекуле, но не открывать ее на новой странице, нажмите на карточке кнопку :





Отображается карточка с информацией для предпросмотра:

Для просмотра молекулы на новой (отдельной) странице левой кнопкой мыши нажмите на карточку молекулы. Появится новая страница с различными информационными блоками:

Структуру молекулы можно сохранить в файл формата .png или mol. Также структуру молекулы и ее свойства можно сохранить в файл формата .pdf.
Чтобы скопировать ссылку на карточку молекулы, нажмите «Скопировать ссылку».
Чтобы скопировать ссылку на карточку молекулы, нажмите «Скопировать ссылку».
3.1.2 Рассчитываемые свойства молекул
Все рассчитываемые свойства молекул содержатся в блоках, описанных ниже.
Чтобы открыть свойства, в левом меню нажмите «Поиск» или «Датасеты», выберите молекулу и нажмите на нее левой кнопкой мыши.
Чтобы открыть свойства, в левом меню нажмите «Поиск» или «Датасеты», выберите молекулу и нажмите на нее левой кнопкой мыши.
Обратите внимание, что если в базе данных есть экспериментальные значения по запрашиваемой молекуле, система выводит именно их, при этом рядом с параметрами отображается зеленый индикатор «EXP». Если экспериментальных данных по молекуле нет, то выводятся прогнозные значения. Это применимо для всех информационных блоков в карточке молекулы.
3.1.2.1 Структурные данные
- Представление в формате канонических SMILES;
- Формат международного химического идентификатора InChI;
- InChI-ключ;
- Название по номенклатуре IUPAC;
- Брутто-формула;
- Молекулярный вес.

Блок содержит информацию о структуре молекулы:
3.1.2.2 Внешние БД
Блок отображается, если для молекулы есть ссылки на внешние базы данных!
Блок содержит список внешних баз данных, в которых присутствует молекула. По ссылкам можно перейти на страницу первоисточника.
Блок содержит список внешних баз данных, в которых присутствует молекула. По ссылкам можно перейти на страницу первоисточника.
3.1.2.3 Синонимы
Блок содержит все известные команде Синтелли синонимы, по которым молекула находится в различных химических базах данных.
3.1.2.4 Физико-химические свойства
Блок содержит физико-химические свойства выбранной молекулы:

1.Water Solubility – растворимость в воде
2.Vapor Pressure – давление насыщенных паров
3.Boiling point – температура кипения (в градусах Цельсия)
4.Flash point – температура вспышки (в градусах Цельсия)
5.Density – плотность
6.Viscosity – вязкость
7.Melting point – температура плавления (в градусах Цельсия)
8.LogP octanol-water – коэффициент распределения соединения в системе октанол-вода
9.Soluble in DMSO – качественный прогноз растворимости в диметилсульфоксиде
2.Vapor Pressure – давление насыщенных паров
3.Boiling point – температура кипения (в градусах Цельсия)
4.Flash point – температура вспышки (в градусах Цельсия)
5.Density – плотность
6.Viscosity – вязкость
7.Melting point – температура плавления (в градусах Цельсия)
8.LogP octanol-water – коэффициент распределения соединения в системе октанол-вода
9.Soluble in DMSO – качественный прогноз растворимости в диметилсульфоксиде

10.Retention time – время удерживания в стандартной хроматографической системе
Условия
Условия

11.Refractive Index – индекс рефракции
3.1.2.5 Биологическая активность
Блок содержит прогноз биологической активности молекулы. В нём доступны модели для расчёта эффективности ингибирования пяти цитохромов: CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4.
Цитохромы P450 — ключевые ферменты, катализирующие метаболизм (окисление) ксенобиотиков в организме. Биологическая функция цитохромов P450 заключается в удалении из организма токсичных соединений. Ингибиторы цитохромов могут стимулировать нежелательные побочные эффекты и приводить к накоплению токсичных соединений в организме. Прогноз ингибирования цитохромов P450 позволяет оценивать метаболизм изучаемых соединений, их безопасность и возможные взаимодействия с другими препаратами.
Цитохромы P450 — ключевые ферменты, катализирующие метаболизм (окисление) ксенобиотиков в организме. Биологическая функция цитохромов P450 заключается в удалении из организма токсичных соединений. Ингибиторы цитохромов могут стимулировать нежелательные побочные эффекты и приводить к накоплению токсичных соединений в организме. Прогноз ингибирования цитохромов P450 позволяет оценивать метаболизм изучаемых соединений, их безопасность и возможные взаимодействия с другими препаратами.
- «Localization» – прогноз органа, где молекулы будут наиболее активны или будут наиболее сконцентрированы;
- «Estimate fraction of metabolized drugs» – предсказание, сколько процентов лекарственного вещества по статистике метаболизируется этим ферментом.
Также блок прогнозирует следующие свойства выбранной молекулы:

При наведении курсора на название цитохрома система отображает всплывающие подсказки по следующим параметрам:
1.Проницаемость через ГЭБ
2.Aromatase
3.Estrogen Receptor Alpha, full length
4.Androgen Receptor, full length
5.Aryl Hydrocarbon Receptor
6.PPAR-gamma
7.Androgen Receptor Ligand-Binding Domain
8.Estrogen Receptor Ligand-Binding Domain
9.p53
10.ATAD5
11.Mitochondrial Membrane Potential
12.SR-ARE (Antioxidant Response Element)
13.Heat Shock Factor Response Element
2.Aromatase
3.Estrogen Receptor Alpha, full length
4.Androgen Receptor, full length
5.Aryl Hydrocarbon Receptor
6.PPAR-gamma
7.Androgen Receptor Ligand-Binding Domain
8.Estrogen Receptor Ligand-Binding Domain
9.p53
10.ATAD5
11.Mitochondrial Membrane Potential
12.SR-ARE (Antioxidant Response Element)
13.Heat Shock Factor Response Element
3.1.2.6 Токсичность
Блок содержит прогнозы опасности и токсичности соединения.
Показатели токсичности разделены на две группы:
Модели летальной дозы
Модели общий токсичности
В рамках первой группы для разных видов животных прогнозируются следующие показатели:
LD50 (median lethal dose) – средняя летальная доза химического вещества, вызывающая гибель 50% животных при введении в организм; выражается в миллиграммах вещества на килограмм массы животного (мг/кг).
LDLo (lowest lethal dose) – наименьшая летальная доза, вызывающая смерть у данного вида животных. Дозировка также указывается на единицу массы тела (мг/кг).
Вторая группа прогнозируемых свойств токсичности состоит из параметров:
Модели летальной дозы
Модели общий токсичности
В рамках первой группы для разных видов животных прогнозируются следующие показатели:
LD50 (median lethal dose) – средняя летальная доза химического вещества, вызывающая гибель 50% животных при введении в организм; выражается в миллиграммах вещества на килограмм массы животного (мг/кг).
LDLo (lowest lethal dose) – наименьшая летальная доза, вызывающая смерть у данного вида животных. Дозировка также указывается на единицу массы тела (мг/кг).
- Мышь перорально LD50 (Mouse Oral LD50)
- Мышь внутривенно LD50 (Mouse Intravenous LD50)
- Мышь интраперитонеально LD50 (Mouse Intraperitoneal LD50)
- Мышь интраперитонеально LDLo (Mouse Intraperitoneal LDLo)
- Кожа мыши LD50 (Mouse Skin LD50) — смертельная доза при нанесении вещества на кожу мышей;
- Мышь подкожно LD50 (Mouse Subcutaneous LD50)
- Мышь внутримышечно LD50 (Mouse Intramuscular LD50)
- Крыса перорально LD50 (Rat Oral LD50)
- Крыса перорально LDLo (Rat Oral LDLo)
- Крыса внутривенно LD50 (Rat Intravenous LD50)
- Крыса интраперитонеально LD50 (Rat Intraperitoneal LD50)
- Крыса накожно LD50 (Rat Skin LD50)
- Крыса подкожно LD50 (Rat Subcutaneous LD50)
- Крыса интраперитонеально LDLo (Rat Intraperitoneal LDLo)
- Гвинейская свинья интраперитонеально LD50 (Guinea Pig Oral LD50)
- Гвинейская свинья интраперитонеально LD50 (Guinea Pig Intraperitoneal LD50)
- Кролик перорально LD50 (Rabbit Oral LD50)
- Кролик внутривенно LD50 (Rabbit Intravenous LD50)
- Кролик внутривенно LDLo (Rabbit Intravenous LDLo)
- Кролик накожно LD50 (Rabbit Skin LD50)
- Собака перорально LD50 (Dog Oral LD50)
- Собака внутривенно LDLo (Dog Intravenous LDLo)
- Собака внутривенно LD50 (Dog Intravenous LD50)
- Кошка внутривенно LD50 (Cat Intravenous LD50)
- Перепел перорально LD50 (Quail Oral LD50)
- Птица дикая перорально LD50 (Bird Wild Oral LD50)
- Курица перорально LD50 (Chicken Oral LD50)
Вторая группа прогнозируемых свойств токсичности состоит из параметров:
- Избирательная токсичность для определенного органа или системы при однократном воздействии (Selective Target-organ Or System Toxicity Single Exposure)
- Острая токсичность при проглатывании (Acute Toxicity Swallowed (ГОСТ - 56957-2016))
- Разъедание глаз (Eye Corrosion)
- Раздражение глаз (Eye Irritation)
- Тератогенность (Developmental Toxicity)
- Репродуктивная токсичность (Reproductive Toxicity)
- Кардиотоксичность (Cardiotoxicity)
- Гепатотоксичность (Hepatotoxicity)
- Канцерогенность (Carcinogenicity)
- DILI (Drug-Induced Liver Injury)
- Тест Эймса

3.1.2.7 Экологические свойства
Блок отображает экологические свойства соединений по нескольким параметрам.
В системе рассчитываются следующие параметры:
- 40 часовой Tetrahymena pyriformis IGC50 – концентрация испытуемого вещества в воде в мг/л, приводящая к 50% ингибированию роста Tetrahymena pyriformis через 40 часов.
- Биоконцентрационный фактор
- 48 hour Daphnia magna LC50 – модель обеспечивает количественный прогноз для Daphnia Magna LC50 (48 часов), выраженный в -log(моль/л) и преобразованный в мг/л.
- «96 часов Fathead Minnow LC50» — концентрация вещества, при которой ожидается гибель 50% популяции Fathead Minnow (вида пресноводных рыб) за 96 часов. Это важный показатель токсичности вещества для рыб, и помогает оценить риски для водных экосистем.
- Acute Aquatic Toxicity (ГОСТ 57455 от 2017 г.)

3.1.2.8 Оценка сложности синтеза
В этом блоке приводится оценка синтетической сложности на основе различных прогностических моделей.
1. «SYBA» — Байесовская оценка синтетической доступности органических соединений.
2. «Complexity (SCScore: Synthetic Complexity Learned from a 12 million Reaction and predicted by neural network)»– шкала Коннора Коли, где «1» означает, что соединение легко синтезируемое, а «5» - сложно синтезируемое.
2. «Complexity (SCScore: Synthetic Complexity Learned from a 12 million Reaction and predicted by neural network)»– шкала Коннора Коли, где «1» означает, что соединение легко синтезируемое, а «5» - сложно синтезируемое.

3.1.2.9 Сходство с лекарственными препаратами
В данном блоке отображаются параметры сходства с лекарственными молекулами.
- Lipinski's rule of five (Правило Липински) – эмпирическое правило, разработанное Липински в 1999 году, помогающее определить, обладает ли химическое соединение нужными значениями физико-химических свойств для достижения высокой биодоступности при пероральном введении;
- Ghose Filter (Фильтр Гоуза) – фильтр, разработанный Гоузом в 1999 году, определяет диапазон значений физико-химических параметров, который отвечает распределению лекарств из базы данных Comprehensive Medicinal Chemistry;
- Oprea's Rule – правило Опреа, разработанное в 1999 году, которое разграничивает потенциальные лекарственные молекулы от нелекарственных молекул;
- Veber's Rule (Правило вебера) – правило о важных физико-химических параметрах для достижения высокой биодоступности при пероральном введении, разработанное в 2002 году Вебером в результате анализа 1100 кандидатов в лекарства;
- QED (quantitative estimate of drug-likeness) – количественная оценка сходства с лекарственными средствами;
- PAINS (pan-assay interference compounds) – фильтр по известным химическим структурам, которые часто дают ложноположительные результаты при высокопроизводительном скрининге.

3.2 Применимость моделей
Для моделей, которые предсказывают биологические, физико-химические, токсикологические, экологические свойства можно включить отображение уровня применимости модели к конкретной, просматриваемой молекуле.
Чтобы отобразить применимость модели, переключите одноименный переключатель, расположенный в правом верхнем углу, на карточке молекулы.
Чтобы отобразить применимость модели, переключите одноименный переключатель, расположенный в правом верхнем углу, на карточке молекулы.
При включении переключателя напротив каждого свойства отображается процент применимости модели к молекуле:
● Если показатель от 0 до 20% - низкая надежность прогнозирования. В тренировочных данных модели было мало молекул, подобных выбранной
● 20-50% Средняя надежность прогнозирования
● 50-100% Высокая надежность прогнозирования
● “Not applicable” модель не может быть применена для прогнозирования свойств этой молекулы
● Если показатель от 0 до 20% - низкая надежность прогнозирования. В тренировочных данных модели было мало молекул, подобных выбранной
● 20-50% Средняя надежность прогнозирования
● 50-100% Высокая надежность прогнозирования
● “Not applicable” модель не может быть применена для прогнозирования свойств этой молекулы


3.3 Поиск по реакциям
В данном разделе можно осуществлять поиск реакций по реактанту, реагенту или итоговому продукту реакции.
Чтобы открыть раздел, в меню слева выберите «Поиск» и нажмите вкладку «По реакциям».
Чтобы открыть раздел, в меню слева выберите «Поиск» и нажмите вкладку «По реакциям».
Молекулярные структуры (реагент, реактант или итоговый продукт) можно искать двумя способами:
● В поисковой строке можно ввести название молекулы в любом удобном формате: SMILES, синонимы, торговое название, код вендора, название в формате IUPAC, CAS-номер и кодовые обозначения из других баз данных.
● В поисковой строке можно ввести название молекулы в любом удобном формате: SMILES, синонимы, торговое название, код вендора, название в формате IUPAC, CAS-номер и кодовые обозначения из других баз данных.


Или
● В молекулярном редакторе нарисовать структурную формулу молекулы. Для это нажмите кнопку «Нарисовать», в появившемся окне нарисуйте молекулу и нажмите «Сохранить».
Дополнительно можно установить следующие параметры поиска в панели «Фильтры» (панель автоматически появляется после первого поиска реакций):
● «Тип соответствия» — фильтр используется для нахождения структур по соответствию молекуле. Например, можно искать точное соответствие структуре или выполнять подструктурный поиск.
● «Роль вещества» — фильтр используется для поиска реакции по роли веществ в реакции, например, можно искать реакции, в которых определенное соединение является продуктом, ректантом или реагентом.
● «Выход, %» — фильтр используется для поиска реакции с определенным процентом выхода продукта.
Для удаления всех настроенных параметров нажмите «Очистить все фильтры».
● В молекулярном редакторе нарисовать структурную формулу молекулы. Для это нажмите кнопку «Нарисовать», в появившемся окне нарисуйте молекулу и нажмите «Сохранить».
Дополнительно можно установить следующие параметры поиска в панели «Фильтры» (панель автоматически появляется после первого поиска реакций):
● «Тип соответствия» — фильтр используется для нахождения структур по соответствию молекуле. Например, можно искать точное соответствие структуре или выполнять подструктурный поиск.
● «Роль вещества» — фильтр используется для поиска реакции по роли веществ в реакции, например, можно искать реакции, в которых определенное соединение является продуктом, ректантом или реагентом.
● «Выход, %» — фильтр используется для поиска реакции с определенным процентом выхода продукта.
Для удаления всех настроенных параметров нажмите «Очистить все фильтры».

В результате система отображает список реакций, подходящий под условия запроса.
Полученные реакции можно отсортировать по релевантности, дате публикации, выходу реакции и количеству шагов.
Полученные реакции можно отсортировать по релевантности, дате публикации, выходу реакции и количеству шагов.

При клике на карточку реакции ее можно открыть в отдельном окне и просмотреть подробную информацию:

3.4 Модуль поиска по литературным источникам
В данном разделе можно осуществлять поиск по научной химической литературе, собранной в базе данных Синтелли.
Чтобы открыть данный раздел, в меню слева выберите «Поиск», далее откройте вкладку «По литературе».
Чтобы открыть данный раздел, в меню слева выберите «Поиск», далее откройте вкладку «По литературе».
Обратите внимание!
- Если в поисковую строку введен какой-либо идентификатор молекулы (SMILES, IUPAC или CAS номер) то будет осуществлен поиск этого соединения в литературе. Также для выполнения поиска по структуре в документах искомое соединение может быть задано через молекулярный редактор:
2. Если в поисковую строку введен текст, то будет осуществлен поиск соответствующего текста в заголовках и аннотациях документов.
Чтобы сразу уточнить поисковый запрос ограничивающими условиями, такие как:
● Авторы,
● DOI,
● Номер патента,
● Аннотация
● Наименование журнала
● Издатель
● Язык
● Дата публикации,
Нажмите “ Добавить условие” под поисковой строкой
Чтобы сразу уточнить поисковый запрос ограничивающими условиями, такие как:
● Авторы,
● DOI,
● Номер патента,
● Аннотация
● Наименование журнала
● Издатель
● Язык
● Дата публикации,
Нажмите “ Добавить условие” под поисковой строкой
Обратите внимание!
Поиск по условиям может быть выполнен, в том числе, при отсутствии запроса в поисковой строке.
Поиск по условиям может быть выполнен, в том числе, при отсутствии запроса в поисковой строке.



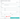

Условия для поиска можно комбинировать. Для этого в открывшемся окне в любом сочетании можно выбрать условие и указать желаемое значение. Для объединения нескольких условий в один поисковый запрос можно применять логические операторы И или ИЛИ. Если выбрано И, оба условия должны войти в запрос, если ИЛИ – одно из условий.
Нажмите кнопку «Подтвердить».
После выполнения поискового запроса его можно уточнить, выбрав фильтры в открывшемся слева окне.
Дополнительно можно установить следующие параметры поиска в панели «Фильтры»:
Нажмите кнопку «Подтвердить».
После выполнения поискового запроса его можно уточнить, выбрав фильтры в открывшемся слева окне.
Дополнительно можно установить следующие параметры поиска в панели «Фильтры»:

«Тип документа» — тип публикации (статья в журнале, патент, обзор, клиническое испытание, статья с конференции);
«Язык» — выбор языка: русский, английский, немецкий;
«Год публикации» — год публикации документа.
Обратите внимание!
Если вы уже указали искомые значение для года публикации ранее, при задании условий поиска, то это поле выбора в фильтрах будет недоступно. Для изменения вам необходимо внести корректировки именно в конструктор запроса.
Для удаления всех настроенных параметров нажмите «Очистить все фильтры».
Для применения фильтров нажмите “Применить”.
После применения всех параметров система скорректирует список найденных публикаций.
«Язык» — выбор языка: русский, английский, немецкий;
«Год публикации» — год публикации документа.
Обратите внимание!
Если вы уже указали искомые значение для года публикации ранее, при задании условий поиска, то это поле выбора в фильтрах будет недоступно. Для изменения вам необходимо внести корректировки именно в конструктор запроса.
Для удаления всех настроенных параметров нажмите «Очистить все фильтры».
Для применения фильтров нажмите “Применить”.
После применения всех параметров система скорректирует список найденных публикаций.
Для найденной статьи вы можете скопировать ссылку, DOI (уникальный идентификатор публикации) или скрыть статью из результатов поиска, нажав на «…» в карточке статьи.

Для отображения полного текста публикации, структур или реакций, которые есть в публикации, нажмите «Читать полностью».
Для просмотра веществ, которые упоминаются в документе нажмите «Структуры», а чтобы открыть реакции нажмите «Реакции».
Кнопки расположены внизу каждой карточки:
Для просмотра веществ, которые упоминаются в документе нажмите «Структуры», а чтобы открыть реакции нажмите «Реакции».
Кнопки расположены внизу каждой карточки:

Для просмотра полной информации о статье нажмите на нее левой клавишей мыши.

4 Раздел «Молекулярный редактор»
Данный раздел позволяет прогнозировать свойства соединений, которых нет в базе данных Синтелли.
Ввести молекулу можно двумя способами:
Подсказка:
Вы можете отобразить структуру уже известной молекулы, внести в нее корректировки в соответствии со своим экспериментом и предсказать свойства нового соединения.
2. Ввести SMILES после нажатия на соответствующую кнопку
Ввести молекулу можно двумя способами:
- Ввести название бренда, название IUPAC, код поставщика или CAS номер в поисковую строку и нажать
Подсказка:
Вы можете отобразить структуру уже известной молекулы, внести в нее корректировки в соответствии со своим экспериментом и предсказать свойства нового соединения.
2. Ввести SMILES после нажатия на соответствующую кнопку
Подсказка: для удаления текстового запроса нажмите на кнопку
3. Нарисовать структуру в молекулярном редакторе.
Для показа свойств нарисованной молекулы нажмите кнопку
страницы молекулярного редактора.
Отобразится карточка молекулы с ее рассчитанными свойствами:
Для показа свойств нарисованной молекулы нажмите кнопку
страницы молекулярного редактора.
Отобразится карточка молекулы с ее рассчитанными свойствами:





в правом верхнем углу

Для экспорта структуры молекулы в файл формата .png нажмите «Скачать»

Для экспорта молекулы и всех рассчитанных свойств в файл формата .pdf нажмите «Скачать PDF».
Чтобы вернуться в молекулярный редактор, нажмите кнопку
Подробное описание всех рассчитанных свойств содержится в главе Рассчитываемые свойства молекул.
Чтобы вернуться в молекулярный редактор, нажмите кнопку
Подробное описание всех рассчитанных свойств содержится в главе Рассчитываемые свойства молекул.

в правом верхнем углу страницы.
5 Раздел «Датасеты»
Данный раздел предназначен для работы с личными датасетами пользователя, а также с тематическими датасетами, предоставленными командой Синтелли.
Чтобы открыть раздел, в меню слева нажмите «Датасеты», далее выберите вкладку «Тематические» или «Личные».
Чтобы открыть раздел, в меню слева нажмите «Датасеты», далее выберите вкладку «Тематические» или «Личные».

5.1 Создание датасета
Находясь в разделе “Датасеты”, нажмите кнопку


Далее введите название и описание датасета и нажмите «Создать»:

После создания датасета он появляется в разделе «Датасеты» во вкладке “Личные”.
5.2 Добавление молекул в датасет
Для добавления молекулы в датасет в меню слева выберите раздел «Датасеты» > вкладку «Тематические» или «Личные» и нажмите кнопку «Плюс» вверху страницы или, если датасет был создан только что, нажмите кнопку «Добавить молекулу» в середине страницы.
Далее из раскрывающегося списка необходимо выбрать метод загрузки молекулы:


● «SMILES» — после выбора данной опции необходимо ввести SMILES в появившееся поле;
● «Молекулярный редактор» — после выбора данной опции отображается молекулярный редактор, где можно нарисовать требуемую структуру;
● «Загрузить из файла» — после выбора данной опции необходимо нажать кнопку
«Выберите файл» и загрузить файл формата .sdf, .csv, .smi;
Обратите внимание!
Загружаемый файл должен иметь колонку, содержащую SMILES соединений. При загрузке вам будет предложено по названиям колонок вашего файла выбрать нужную колонку.
● «Поиск по синонимам» — после выбора данной опции необходимо ввести синоним (название молекулы) в появившемся поле.
Найденная молекула отображается в окне предпросмотра. Чтобы добавить ее в датасет, нажмите кнопку «Загрузить»:
● «Молекулярный редактор» — после выбора данной опции отображается молекулярный редактор, где можно нарисовать требуемую структуру;
● «Загрузить из файла» — после выбора данной опции необходимо нажать кнопку
«Выберите файл» и загрузить файл формата .sdf, .csv, .smi;
Обратите внимание!
Загружаемый файл должен иметь колонку, содержащую SMILES соединений. При загрузке вам будет предложено по названиям колонок вашего файла выбрать нужную колонку.
● «Поиск по синонимам» — после выбора данной опции необходимо ввести синоним (название молекулы) в появившемся поле.
Найденная молекула отображается в окне предпросмотра. Чтобы добавить ее в датасет, нажмите кнопку «Загрузить»:

5.3 Просмотр данных о молекуле в личном датасете
Чтобы просмотреть молекулы нажмите на датасет левой кнопкой мыши. Работа с молекулами в личном датасете осуществляется также, как и в публичной базе данных.
5.4 Выделение датасета
Для выделения / снятия выделения с датасета необходимо нажать кнопку и выбрать команду «Выделить»:
Далее из раскрывающегося списка необходимо выбрать метод загрузки молекулы.

5.5 Редактирование датасета
Для редактирования названия и описания датасета необходимо нажать кнопку
При редактировании можно изменить название и описание датасета.
и выбрать команду «Редактировать»:

5.6 Просмотр сообщений журнала
Для просмотра сообщений журнала необходимо нажать кнопку
При редактировании в журнале отображаются данные об изменении датасета, даты изменения и другая информация: нии можно изменить название и описание датасета.
«Просмотр сообщений журнала»:
на датасете и выбрать команду


5.7 Удаление датасета
Нажмите кнопку
и в появившемся меню выберите «Удалить»:



5.8 Копирование датасета
Чтобы скопировать датасет со всеми молекулами, выделите датасет. Чтобы выделить датасет, зажмите клавишу Ctrl и кликните на нужный датасет (для выделения по одному) или Shift (для выделения группы дасатетов), после этого нажмите кнопку


5.9 Объединение датасетов
Чтобы объединить датасеты в один, выделите два или более датасетов и нажмите кнопку


5.10 Показ датасета на SynMap
Чтобы показать датасет на координатной плоскости SynMap, выделите датасет и нажмите кнопку


5.11 Экспорт датасета
Чтобы экспортировать датасет со всеми молекулами и их свойствами, выделите датасет и нажмите кнопку


Если свойства молекул не рассчитаны, система спрашивает, надо ли рассчитать все свойства или экспортировать только SMILES:

Экспорт может осуществляться в форматы .CSV или .SDF:

Файл загружается на локальный диск персонального компьютера.
5.12 Расчет свойств молекулы
Чтобы рассчитать свойства соединений, входящих в выделенный датасет (токсикологические, физико-химические, биологические и проч.), выделите датасет и нажмите кнопку






6 Раздел «SynMap»
Данный раздел позволяет наглядно проецировать структуры молекул на двумерной плоскости так, чтобы молекулы, близкие по свойствам, оказывались рядом, образуя кластеры.
Дополнительно можно генерировать химические соединения с заданными свойствами (QED, Boiling point, Melting point, Mouse oral LD50, LogP, LogS, DMSO Solubility, Brutto, MMap).
Подсказки:
- Степень прозрачности точек можно регулировать бегунком в правом верхнем углу.
- Карту можно приблизить или отдалить при помощи прокрутки колеса мыши, либо с помощью кнопок увеличения масштаба. Для того, чтобы вернуться в первоначальное состояние карты (по умолчанию), нажмите на кнопку (автоматические масштабирование).
- Для перемещения всей карты при помощи мыши, нажмите
6.1 Просмотр групп химических соединений
Раздел позволяет получить быстрое и наглядное представление об основных группах химических соединений, которые есть в датасете. Модель производит проецирование структур химических соединений в координаты X и Y на двумерной плоскости. Сами по себе координаты не несут физического смысла, однако модель настроена таким образом, что структурно близкие соединения оказываются рядом.
В итоге на карте химические соединения распределяются так, что можно увидеть знакомые кластеры: кластер простых линейных алифатических соединений, кластер стероидов, бисфенилов, психоактивных веществ и т.п.
Данный раздел можно открыть двумя способами:
1. В разделе «Датасеты» выбрать подготовленный датасет и показать его на SynMap.
-ИЛИ-
2. Открыть раздел «SynMap» и, чтобы отобразить соединения на карте нажать кнопку «+» в блоке «Слои».
В итоге на карте химические соединения распределяются так, что можно увидеть знакомые кластеры: кластер простых линейных алифатических соединений, кластер стероидов, бисфенилов, психоактивных веществ и т.п.
Данный раздел можно открыть двумя способами:
1. В разделе «Датасеты» выбрать подготовленный датасет и показать его на SynMap.
-ИЛИ-
2. Открыть раздел «SynMap» и, чтобы отобразить соединения на карте нажать кнопку «+» в блоке «Слои».

Далее необходимо выбрать заранее подготовленный публичный и / или личный датасет:
Подсказка:
После выбора датасетов нажмите «Выбрать».
Загрузка больших датасетов занимает некоторое время. Если вы хотите убедиться в том, что она все-таки идет, обратите внимание на блок «Слои» в правом верхнем углу, там появится название датасета и индикатор загрузки. По окончании загрузки вы увидите на карте все молекулы.
- Система позволяет выбрать сразу несколько датасетов.
После выбора датасетов нажмите «Выбрать».
Загрузка больших датасетов занимает некоторое время. Если вы хотите убедиться в том, что она все-таки идет, обратите внимание на блок «Слои» в правом верхнем углу, там появится название датасета и индикатор загрузки. По окончании загрузки вы увидите на карте все молекулы.

Каждая точка на этой карте — это молекула, при наведении на точку отображается карточка этой молекулы. Если загружено сразу несколько датасетов, каждый из них отображается отдельным цветом.
По карте можно легко определить, какие основные классы соединений преобладают в этом датасете. Например, в нижней части по центру находится сгущение близких по свойствам молекул. Структуру каждой молекулы можно посмотреть, наведя на нее курсор. В данном случае в этой области расположены простые молекулы:
По карте можно легко определить, какие основные классы соединений преобладают в этом датасете. Например, в нижней части по центру находится сгущение близких по свойствам молекул. Структуру каждой молекулы можно посмотреть, наведя на нее курсор. В данном случае в этой области расположены простые молекулы:

В правой области карты можем увидеть класс стероидных соединений.

На карте можно выделить отдельные группы молекул, а также экспортировать их в отдельный датасет.
Для выделения нужной области нажмите кнопку
Для выделения нужной области нажмите кнопку

или
и мышью выделите прямоугольную или

произвольную область соответственно.
Подсказка:
На этом этапе можно двигать границы выбранной области. Максимально возможное количество молекул для выделения и сохранения – 50 000.
На отдельной панели отображаются структуры, которые входят в выбранную область:
- Для отмены выделения необходимо дважды кликнуть левой кнопкой мыши за пределами выделенной области.
На этом этапе можно двигать границы выбранной области. Максимально возможное количество молекул для выделения и сохранения – 50 000.
На отдельной панели отображаются структуры, которые входят в выбранную область:

При нажатии на кнопку

можно просмотреть информацию о молекуле, скопировать SMILES, открыть
в молекулярном редакторе, скачать структуру в формате .png, или добавить молекулу в другой датасет. Более подробно информация по этим действиям описана в разделе Работа с молекулярными структурами.
Выбранные молекулы можно также сохранить в новом или уже созданном датасете.
Чтобы сохранить структуру в имеющийся датасет, на выбранной структуре нажмите кнопку «Сохранить в датасет», выберите в списке сет и нажмите кнопку «Добавить в датасет»:
Выбранные молекулы можно также сохранить в новом или уже созданном датасете.
Чтобы сохранить структуру в имеющийся датасет, на выбранной структуре нажмите кнопку «Сохранить в датасет», выберите в списке сет и нажмите кнопку «Добавить в датасет»:
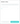
Чтобы сохранить структуру в новый датасет, введите название нового сета, нажмите кнопку «Создать новый».
После того, как новый датасет появится в списке датасетов, кликнете на него, произведя его выбор и нажмите «Добавить в датасет»:
Сохраненный сет можно найти в разделе «Датасеты».
При помощи кнопки




можно включить или выключить отображение датасета на карте.
Чтобы удалить датасет с SynMap, нажмите кнопку
Молекулы, размещенные на карте, можно отобразить на 3D-плоскости. Для этого нажмите на соответствующую кнопку в правом верхнем углу карты.


Подсказка:
- Для возможности отображения датасета на 3D плоскости происходит расчет его трехмерных координат, поэтому отображение больших датасетов может занимать значительное время.
6.2 Генерация соединений с заданными свойствами
Чтобы создать новые соединения, в разделе «SynMap» и нажмите на «+» в блоке «Генераторы».

В открывшемся окне можно задать:

● «Название» (произвольное);
● Родительскую связь (для повторных генераций);
● «Эпохи» — количество попыток системы придумать новую молекулу; при увеличении количества эпох нейронная сеть будет иметь больше времени на генерацию соединений, косвенно это повлияет на качество генерации (чем больше эпох, тем больше будет предложено вариантов), но стоит учесть, что время генерации увеличивается пропорционально;
● Свойство, по которому будут создаваться соединения. Одновременно можно выбирать сразу несколько свойств:
«MMap» (Molecular Map) — применяется для генерации молекул с определенными структурными или функциональными характеристиками из какого-либо участка карты. При выборе данного свойства необходимо ввести координаты и радиус, указать референсное соединение в строку SMILES, либо выбрать место прямо на карте;
Обратите внимание!
Чтобы воспользоваться генерацией молекул из участка карты необходимо предварительно отобразить интересующий датасет на карте .
● Родительскую связь (для повторных генераций);
● «Эпохи» — количество попыток системы придумать новую молекулу; при увеличении количества эпох нейронная сеть будет иметь больше времени на генерацию соединений, косвенно это повлияет на качество генерации (чем больше эпох, тем больше будет предложено вариантов), но стоит учесть, что время генерации увеличивается пропорционально;
● Свойство, по которому будут создаваться соединения. Одновременно можно выбирать сразу несколько свойств:
«MMap» (Molecular Map) — применяется для генерации молекул с определенными структурными или функциональными характеристиками из какого-либо участка карты. При выборе данного свойства необходимо ввести координаты и радиус, указать референсное соединение в строку SMILES, либо выбрать место прямо на карте;
Обратите внимание!
Чтобы воспользоваться генерацией молекул из участка карты необходимо предварительно отобразить интересующий датасет на карте .

«QED» (Quantitative Estimate of Drug-likeness) — применяется для генерации молекул, которые имеют высокую вероятность быть эффективными и безопасными лекарственными средствами. QED оценивает, насколько химическая структура соединения соответствует типичным характеристикам успешных лекарств.

«Boiling point» (температура кипения) — применяется для создания молекулы с предопределенной температурой перехода из жидкого состояния в газообразное. Это свойство важно для применений, где температурная стабильность и летучесть играют ключевую роль.
«Melting point» (точка плавления) — применяется для создания молекул с конкретной температурой перехода из твердого состояния в жидкое. Это свойство критично для веществ, предназначенных для использования в определенных температурных условиях.
«Mouse oral LD50» (Мышь перорально LD50) — применяется для создания молекулы с заданным уровнем токсичности при пероральном введении, что важно для оценки безопасности новых соединений.
«LogP» — применяется для создания молекул с определенной липофильностью, что влияет на их способность проникать через биологические мембраны и распределяться в организме.
«LogS» — применяется для создания молекулы с предварительно заданной растворимостью в воде, ключевым фактором для биодоступности и применения вещества.
«DMSO solubility» (Растворимость в ДМСО) — применяется для создания молекул, которые хорошо растворяются в диметилсульфоксиде, что важно для многих лабораторных исследований и применений.
«Brutto» — применяется для создания молекулы с определенным химическим составом, что является основой для идентификации и классификации химических соединений.
«Melting point» (точка плавления) — применяется для создания молекул с конкретной температурой перехода из твердого состояния в жидкое. Это свойство критично для веществ, предназначенных для использования в определенных температурных условиях.
«Mouse oral LD50» (Мышь перорально LD50) — применяется для создания молекулы с заданным уровнем токсичности при пероральном введении, что важно для оценки безопасности новых соединений.
«LogP» — применяется для создания молекул с определенной липофильностью, что влияет на их способность проникать через биологические мембраны и распределяться в организме.
«LogS» — применяется для создания молекулы с предварительно заданной растворимостью в воде, ключевым фактором для биодоступности и применения вещества.
«DMSO solubility» (Растворимость в ДМСО) — применяется для создания молекул, которые хорошо растворяются в диметилсульфоксиде, что важно для многих лабораторных исследований и применений.
«Brutto» — применяется для создания молекулы с определенным химическим составом, что является основой для идентификации и классификации химических соединений.

«Complexity» — Этот параметр отражает структурную сложность молекулы. Чем выше значение сложности, тем более сложные и, многофункциональные генерируются молекулы. Низкая сложность указывает на более простые, базовые структуры.
«SYBA» — Этот параметр относится к вероятностной оценке синтезируемости молекул. Он предсказывает насколько легко или сложно будет синтезировать предложенную молекулярную структуру в лабораторных условиях.
Для каждого свойства отображается ползунок. Он отвечает за степень качественного соответствия созданного массива молекул заданным условиям. Если ползунок будет установлен максимально вправо, то массив соединений будет сгенерирован с меньшим количеством молекул, но максимально точно отвечающий запросу (например, при создании соединений с низкой токсичностью на карте останутся молекулы с самой низкой токсичностью).
Чем ближе ползунок к левому краю, тем больше будет сгенерированных вариантов, но они с меньшей точностью будут соответствовать заданному запросу.
«SYBA» — Этот параметр относится к вероятностной оценке синтезируемости молекул. Он предсказывает насколько легко или сложно будет синтезировать предложенную молекулярную структуру в лабораторных условиях.
Для каждого свойства отображается ползунок. Он отвечает за степень качественного соответствия созданного массива молекул заданным условиям. Если ползунок будет установлен максимально вправо, то массив соединений будет сгенерирован с меньшим количеством молекул, но максимально точно отвечающий запросу (например, при создании соединений с низкой токсичностью на карте останутся молекулы с самой низкой токсичностью).
Чем ближе ползунок к левому краю, тем больше будет сгенерированных вариантов, но они с меньшей точностью будут соответствовать заданному запросу.
Для удаления ненужного свойства нажмите

После нажатия на кнопку «Создать» начнется генерация соединений, постепенно на карте начнут появляться новые молекулы, окрашенные в другой цвет.

Можно настроить любой цвет для молекул, нажав на квадрат с цветом:
Сгенерированные молекулы также можно экспортировать в отдельный датасет. Для этого выделите нужную область и экспортируйте полученные данные, нажав на кнопку «Сохранить в датасет».
Система предложит вам создать новый датасет или добавить их в существующий.
Датасет с новыми молекулами появится в разделе «Датасеты».
Подсказка:
Если датасет появился, а молекул в нем нет, нажмите на кнопку
Система предложит вам создать новый датасет или добавить их в существующий.
Датасет с новыми молекулами появится в разделе «Датасеты».
Подсказка:
Если датасет появился, а молекул в нем нет, нажмите на кнопку


Информация по работе с датасетом (изменение названия, выделение, удаление и проч.) находится в разделе «Датасеты».
7 Раздел «Предсказание реакции»
Раздел экспериментальный!
Раздел посвящен планированию синтеза химических соединений и предсказанию путей химических превращений. В этом разделе можно проводить следующие виды прогнозирования:
1. Синтез — предсказывание возможных продуктов реакции на основе реагентов, вступающих в нее.
2. Ретросинтез — предсказывание реагентов для получения запрашиваемого соединения. На данный момент доступен одностадийный прогноз, в будущем планируется доработать эту опцию до полноценного ретро синтетического анализа.
Раздел посвящен планированию синтеза химических соединений и предсказанию путей химических превращений. В этом разделе можно проводить следующие виды прогнозирования:
1. Синтез — предсказывание возможных продуктов реакции на основе реагентов, вступающих в нее.
2. Ретросинтез — предсказывание реагентов для получения запрашиваемого соединения. На данный момент доступен одностадийный прогноз, в будущем планируется доработать эту опцию до полноценного ретро синтетического анализа.
7.1 Синтез
Для прогнозирования возможных продуктов реакции на основе реагентов, вступающих в нее, откройте раздел «Предсказание реакции», выберите вкладку «Синтез» и нажмите «+»:

Для прогнозирования возможных продуктов реакции на основе реагентов, вступающих в нее, откройте раздел «Предсказание реакции», выберите вкладку «Синтез» и нажмите «+»:

● «SMILES» — при выборе данной опции появляется поле для ввода SMILES.
● «Молекулярный редактор» — при выборе данной опции появляется окно молекулярного редактора, где можно нарисовать химическую структуру.
● «Поиск по синонимам» — при выборе данной опции появится поле для ввода синонимов.
После появления молекулы в окне предпросмотра нажмите «Загрузить».
Чтобы смоделировать исход реакции, нужно добавить по меньшей мере 2 молекулы.
Если вы хотите добавить агент, то вам следует добавить его в этом месте:
● «Молекулярный редактор» — при выборе данной опции появляется окно молекулярного редактора, где можно нарисовать химическую структуру.
● «Поиск по синонимам» — при выборе данной опции появится поле для ввода синонимов.
После появления молекулы в окне предпросмотра нажмите «Загрузить».
Чтобы смоделировать исход реакции, нужно добавить по меньшей мере 2 молекулы.
Если вы хотите добавить агент, то вам следует добавить его в этом месте:

Подсказки:
- При необходимости можно удалить или изменить молекулу в графическом редакторе, подведя курсор к карточке и нажав соответствующую кнопку:

- Для удаления всех молекул на странице нажмите кнопку «Сбросить».
После ввода молекул нажмите «Предсказать».
Как мы видим, первый ответ с надежностью прогноза 0.97 корректный:

В позициях с надежностью меньше 0.96 можно увидеть неожиданные результаты, порой модель генерирует даже алхимические преобразования.
Это не ошибка, а особенности нейросети типа Transformer.
Это не ошибка, а особенности нейросети типа Transformer.
7.2 Ретросинтез
Для получения искомого соединения откройте раздел «Предсказание реакции» и выберите вкладку «Ретросинтез». Далее нажмите «+»:
Далее выберите способ ввода молекулы:
● «SMILES» — при выборе данной опции появляется поле для ввода SMILES;
● «Молекулярный редактор» — при выборе данной опции появляется окно молекулярного редактора, где можно нарисовать химическую структуру;
● «Поиск по синонимам» — при выборе данной опции появится поле для ввода синонимов.
После появления молекулы в окне предпросмотра нажмите «Загрузить».
Подсказки:
● «Молекулярный редактор» — при выборе данной опции появляется окно молекулярного редактора, где можно нарисовать химическую структуру;
● «Поиск по синонимам» — при выборе данной опции появится поле для ввода синонимов.
После появления молекулы в окне предпросмотра нажмите «Загрузить».
Подсказки:
- При необходимости можно удалить или изменить молекулу в графическом редакторе, подведя курсор к карточке и нажав соответствующую кнопку:
Для удаления всех молекул на странице нажмите кнопку «Сбросить».
После ввода молекул нажмите «Предсказать».
После ввода молекул нажмите «Предсказать».



8 Раздел “Спектры”
Данный раздел предсказывает 3 вида спектров:
1) Модуль ядерного магнитного резонанса (ЯМР) позволяет прогнозировать спектральные данные ЯМР (13C, 1H и другие ядра) для малых органических молекул. Результат представлен в виде набора "интенсивность - химический сдвиг". Для спектров 1H также прогнозируется мультиплетность.
2) Модуль предсказания масс-спектров QToF MS/MS единичного иона с точной массой. Спектры вычисляются для низкого (10 эВ), среднего (20 эВ) и высокого (40 эВ) уровней энергии. Результат представлен в виде набора пар “точная масса иона – относительная интенсивность”.
3) Модуль ИК-спектроскопии позволяет прогнозировать ИК-спектр для малых органических молекул при различных вариантах регистрации (газовая фаза, KBr и т.д.). Результат отображается в виде непрерывного графика в осях "длина волны" (в нм) и "относительная интенсивность".
1) Модуль ядерного магнитного резонанса (ЯМР) позволяет прогнозировать спектральные данные ЯМР (13C, 1H и другие ядра) для малых органических молекул. Результат представлен в виде набора "интенсивность - химический сдвиг". Для спектров 1H также прогнозируется мультиплетность.
2) Модуль предсказания масс-спектров QToF MS/MS единичного иона с точной массой. Спектры вычисляются для низкого (10 эВ), среднего (20 эВ) и высокого (40 эВ) уровней энергии. Результат представлен в виде набора пар “точная масса иона – относительная интенсивность”.
3) Модуль ИК-спектроскопии позволяет прогнозировать ИК-спектр для малых органических молекул при различных вариантах регистрации (газовая фаза, KBr и т.д.). Результат отображается в виде непрерывного графика в осях "длина волны" (в нм) и "относительная интенсивность".
8.1 Ядерно-магнитный резонанс
Для предсказания спектров ЯМР необходимо открыть раздел «Спектры» и нажать на вкладку «Ядерно-магнитный резонанс».
На открывшейся странице:
На открывшейся странице:

● В поле «Исходная составная структура» введите InСhI или SMILES;
–ИЛИ–
● Нажмите «Нарисовать молекулу» (в молекулярном редакторе нарисуйте молекулу).
Далее выберите метод ЯМР (1H, 13C, 15N, 19F) и нажмите
–ИЛИ–
● Нажмите «Нарисовать молекулу» (в молекулярном редакторе нарисуйте молекулу).
Далее выберите метод ЯМР (1H, 13C, 15N, 19F) и нажмите

Обозначения 1H, 13C, 15N, 19F относятся к конкретным изотопам элементов, которые можно спрогнозировать:
● 1H (Протий, Водород):
При выборе этого параметра, модуль предсказывает ЯМР-спектр на основе самого распространенного изотопа водорода (1H). Это позволяет анализировать и интерпретировать химическое окружение водородных атомов в молекуле, что является ключевым для понимания органических соединений.
● 13C (Углерод):
Выбрав этот параметр, пользователи получат предсказания ЯМР-спектров для изотопа углерода 13C. Это помогает исследовать структуру углеродного скелета молекул, обеспечивая важную информацию о строении органических соединений.
● 15N (Азот):
Этот выбор позволяет модулю сгенерировать предсказания ЯМР-спектров для изотопа азота 15N. Это особенно полезно для анализа азотсодержащих соединений, включая аминокислоты и белки, улучшая понимание их молекулярной структуры.
● 19F (Фтор):
При выборе фтора 19F, модуль предоставляет предсказания ЯМР-спектров для этого изотопа. Это ценно при исследовании фторсодержащих соединений, которые широко используются в фармацевтике и органической химии, предоставляя детальную информацию о окружении атомов фтора.
● 1H (Протий, Водород):
При выборе этого параметра, модуль предсказывает ЯМР-спектр на основе самого распространенного изотопа водорода (1H). Это позволяет анализировать и интерпретировать химическое окружение водородных атомов в молекуле, что является ключевым для понимания органических соединений.
● 13C (Углерод):
Выбрав этот параметр, пользователи получат предсказания ЯМР-спектров для изотопа углерода 13C. Это помогает исследовать структуру углеродного скелета молекул, обеспечивая важную информацию о строении органических соединений.
● 15N (Азот):
Этот выбор позволяет модулю сгенерировать предсказания ЯМР-спектров для изотопа азота 15N. Это особенно полезно для анализа азотсодержащих соединений, включая аминокислоты и белки, улучшая понимание их молекулярной структуры.
● 19F (Фтор):
При выборе фтора 19F, модуль предоставляет предсказания ЯМР-спектров для этого изотопа. Это ценно при исследовании фторсодержащих соединений, которые широко используются в фармацевтике и органической химии, предоставляя детальную информацию о окружении атомов фтора.
Подсказки:
- Если необходимо изменить структуру, нажмите кнопку
- При необходимости удалить все выставленные параметры, нажмите



Каждый атом в молекуле пронумерован.
Полученный спектр отображается в поле «Вычисленные результаты», позволяя пользователю наглядно увидеть характерные пики и их распределение.
В поле «Вычисленные результаты» отображается спектр, и для каждого пронумерованного атома в таблице рассчитывается мультиплетность («Multiplet») и химический сдвиг («Chemical shift»). Мультиплетность (Multiplet): Для каждого атома рассчитывается мультиплетность, которая описывает шаблон расщепления пика в спектре. Мультиплетность указывает на количество соседних атомов водорода (или других ядер), влияющих на данный атом, и может быть синглетом, дублетом, триплетом и т.д.
Химический Сдвиг (Meas. Shift): Для каждого атома определяется химический сдвиг, который отражает его электронное окружение. Химический сдвиг измеряется в частотных единицах (обычно в ppm - частях на миллион) и предоставляет информацию о типе химической связи и электронной структуре молекулы.
Количество строк в таблице зависит от количества атомов.
Полученный спектр отображается в поле «Вычисленные результаты», позволяя пользователю наглядно увидеть характерные пики и их распределение.
В поле «Вычисленные результаты» отображается спектр, и для каждого пронумерованного атома в таблице рассчитывается мультиплетность («Multiplet») и химический сдвиг («Chemical shift»). Мультиплетность (Multiplet): Для каждого атома рассчитывается мультиплетность, которая описывает шаблон расщепления пика в спектре. Мультиплетность указывает на количество соседних атомов водорода (или других ядер), влияющих на данный атом, и может быть синглетом, дублетом, триплетом и т.д.
Химический Сдвиг (Meas. Shift): Для каждого атома определяется химический сдвиг, который отражает его электронное окружение. Химический сдвиг измеряется в частотных единицах (обычно в ppm - частях на миллион) и предоставляет информацию о типе химической связи и электронной структуре молекулы.
Количество строк в таблице зависит от количества атомов.
8.2 Масс-спектрометрия
Для предсказания масс-спектров необходимо открыть раздел «Спектры» и нажать на вкладку «Масс-спектрометрия».
1. На открывшейся странице:
● В поле «Исходная составная структура» введите InСhI или SMILES;
–ИЛИ–
● Нажмите «Нарисовать молекулу» (в молекулярном редакторе нарисуйте молекулу).
2. Далее установите параметры для создания спектра:
● «Спектральный тип» — тип масс-спектра. Это может быть полный спектр, фрагментационный спектр или другие специализированные типы спектров, каждый из которых предоставляет различные сведения о молекуле.
● «Ионный режим» (Positive или Negative) — указывает, будет ли спектр сгенерирован для положительно и отрицательно заряженных ионов. Положительный режим используется для поиска молекул, которые легко образуют катионы, а отрицательный режим предназначен для молекул, которые образуют анионы.
● «Тип аддукта» — тип аддукта важен для определения молекулярного веса целевого соединения. Аддукты возникают, когда ион образует связь с другими веществами, такими как растворители или контраионы.
● «Округление RI» (Retention Index) — степень округления индекса удерживания в спектре. Индекс удерживания помогает в идентификации соединений путем сравнения времени их удерживания в колонке хроматографа со временем удерживания известных стандартов.
● «Округление m/z» (mass-to-charge ratio) — параметр определяет, как будет округляться отношение массы иона к его заряду в спектре. Это важно для точности идентификации молекул и сравнения с известными стандартами.
После выбора нужных параметров нажмите кнопку
● В поле «Исходная составная структура» введите InСhI или SMILES;
–ИЛИ–
● Нажмите «Нарисовать молекулу» (в молекулярном редакторе нарисуйте молекулу).
2. Далее установите параметры для создания спектра:
● «Спектральный тип» — тип масс-спектра. Это может быть полный спектр, фрагментационный спектр или другие специализированные типы спектров, каждый из которых предоставляет различные сведения о молекуле.
● «Ионный режим» (Positive или Negative) — указывает, будет ли спектр сгенерирован для положительно и отрицательно заряженных ионов. Положительный режим используется для поиска молекул, которые легко образуют катионы, а отрицательный режим предназначен для молекул, которые образуют анионы.
● «Тип аддукта» — тип аддукта важен для определения молекулярного веса целевого соединения. Аддукты возникают, когда ион образует связь с другими веществами, такими как растворители или контраионы.
● «Округление RI» (Retention Index) — степень округления индекса удерживания в спектре. Индекс удерживания помогает в идентификации соединений путем сравнения времени их удерживания в колонке хроматографа со временем удерживания известных стандартов.
● «Округление m/z» (mass-to-charge ratio) — параметр определяет, как будет округляться отношение массы иона к его заряду в спектре. Это важно для точности идентификации молекул и сравнения с известными стандартами.
После выбора нужных параметров нажмите кнопку
Подсказки:
- Если необходимо изменить структуру, нажмите кнопку
- При необходимости удалить все выбранные параметры нажмите


После окончания вычислений на странице отобразятся следующие спектры:

● «Изотопное распределение» — отображает распределение различных изотопов элементов в молекуле. Изотопное распределение важно для точной идентификации и характеристики молекул, поскольку разные изотопы одного и того же элемента могут влиять на массу и структуру молекулы.
● «LE MsMs Spectrum (10V), [M+H]+» — отображает масс-спектрометрию низкой энергии (LE) при 10 вольтах для иона [M+H]+.
● «ME MsMs Spectrum (20V), [M+H]+» — отображает масс-спектрометрию средней энергии (ME) при 20 вольтах для иона [M+H]+.
● «HE MsMs Spectrum (40V), [M+H]+» — отображает масс-спектрометрию высокой энергии (HE) при 40 вольтах для иона [M+H]+.
● «LE MsMs Spectrum (10V), [M+H]+» — отображает масс-спектрометрию низкой энергии (LE) при 10 вольтах для иона [M+H]+.
● «ME MsMs Spectrum (20V), [M+H]+» — отображает масс-спектрометрию средней энергии (ME) при 20 вольтах для иона [M+H]+.
● «HE MsMs Spectrum (40V), [M+H]+» — отображает масс-спектрометрию высокой энергии (HE) при 40 вольтах для иона [M+H]+.
8.3 Инфракрасная спектрометрия
Чтобы предсказать ИК-спектр, откройте раздел «Спектры» и нажмите на вкладку «Инфракрасная спектрометрия».
1. На открывшейся странице:
● В поле «Исходная составная структура» введите InСhI или SMILES;
–ИЛИ–
● Нажмите «Нарисовать молекулу» (в молекулярном редакторе нарисуйте молекулу).
2. Выберите «Метод съемки»:
● «gas (газовая фаза)» — метод подразумевает измерение ИК-спектра вещества в газовой фазе. Использование газовой фазы предпочтительно для получения чистых и неперекрывающихся спектральных линий. Этот метод подходит для газообразных образцов или для тех, которые можно испарить без разложения.
● «liquid (жидкая фаза)» — в этом методе ИК-спектр снимается для образца в жидкой фазе. Он подходит для веществ, находящихся в жидком состоянии при комнатной температуре или для тех, которые могут быть расплавлены. Жидкие образцы могут требовать использования специальных кювет.
● «CCl4» — ИК-спектроскопия с использованием растворителя тетрахлорметана (CCl4). Подходит для веществ, растворимых в CCl4, используются преимущественно для анализа образцов, которые могут взаимодействовать или разлагаться в других растворителях.
● «KBr (бромид калия)» — используется для создания таблеток из порошка образца, смешанного с KBr. Этот метод хорошо подходит для анализа твердых образцов, которые сложно испарить или растворить.
● «nujolmull (суспензия в вазелиновом масле )» — образец смешивается с минеральным маслом для формирования пасты. Этот метод подходит для твердых веществ, которые трудно или невозможно растворить и которые не могут быть анализированы в виде KBr таблетки.
После выбора нужных параметров нажмите кнопку
● В поле «Исходная составная структура» введите InСhI или SMILES;
–ИЛИ–
● Нажмите «Нарисовать молекулу» (в молекулярном редакторе нарисуйте молекулу).
2. Выберите «Метод съемки»:
● «gas (газовая фаза)» — метод подразумевает измерение ИК-спектра вещества в газовой фазе. Использование газовой фазы предпочтительно для получения чистых и неперекрывающихся спектральных линий. Этот метод подходит для газообразных образцов или для тех, которые можно испарить без разложения.
● «liquid (жидкая фаза)» — в этом методе ИК-спектр снимается для образца в жидкой фазе. Он подходит для веществ, находящихся в жидком состоянии при комнатной температуре или для тех, которые могут быть расплавлены. Жидкие образцы могут требовать использования специальных кювет.
● «CCl4» — ИК-спектроскопия с использованием растворителя тетрахлорметана (CCl4). Подходит для веществ, растворимых в CCl4, используются преимущественно для анализа образцов, которые могут взаимодействовать или разлагаться в других растворителях.
● «KBr (бромид калия)» — используется для создания таблеток из порошка образца, смешанного с KBr. Этот метод хорошо подходит для анализа твердых образцов, которые сложно испарить или растворить.
● «nujolmull (суспензия в вазелиновом масле )» — образец смешивается с минеральным маслом для формирования пасты. Этот метод подходит для твердых веществ, которые трудно или невозможно растворить и которые не могут быть анализированы в виде KBr таблетки.
После выбора нужных параметров нажмите кнопку
Подсказки:
- Если необходимо изменить структуру, нажмите кнопку
- При необходимости удалить все выбранные параметры нажмите
На странице отобразятся вычисленные спектры и нумерация атомов в молекуле:


9 Раздел «Стоимость синтеза»
Данный раздел позволяет спрогнозировать стоимость конечных синтетических соединений.
Чтобы рассчитать данные, откройте раздел «Стоимость синтеза».
Чтобы рассчитать данные, откройте раздел «Стоимость синтеза».
1. Введите молекулу, которую необходимо получить. Для этого в поле «Продукт» можно ввести InChI или SMILES (или добавить молекулу при помощи молекулярного редактора).
2. При желании добавьте реагент в поле «Реагент», введя InChI или SMILES (или добавив молекулу при помощи молекулярного редактора).
3. В поле «Вес в граммах» введите желаемый вес синтезируемого вещества в граммах.
4. В поле «Количество стадий (от… до…)» введите количество этапов синтезирования продукта (возможно ввести не более 6 стадий).
После выбора нужных параметров нажмите кнопку
2. При желании добавьте реагент в поле «Реагент», введя InChI или SMILES (или добавив молекулу при помощи молекулярного редактора).
3. В поле «Вес в граммах» введите желаемый вес синтезируемого вещества в граммах.
4. В поле «Количество стадий (от… до…)» введите количество этапов синтезирования продукта (возможно ввести не более 6 стадий).
После выбора нужных параметров нажмите кнопку
Подсказки:
- Если необходимо изменить структуру, нажмите кнопку «Изменить»
- При необходимости удалить все выбранные параметры нажмите «Сбросить»
- Если соединение содержит стереохимию в SMILES и алгоритм не находит пути синтеза, рекомендуется удалить стереохимию из SMILES. Возможно, будут найдены пути синтеза, подходящие для интересующего вещества.
- Для промышленного синтеза рекомендуется вводить вес от 100г.
- Для первоначальной оценки возможности синтеза максимальное число стадий рекомендуется поставить 6, а минимальное 1. Возможно, скорость выполнения будет дольше, но шанс найти реакции выше. Далее уже уменьшать максимальное количество стадий или увеличивать минимальное.
- Не рекомендуется ставить для мин и макс значения количества стадий одно и то же число, например, мин=3 макс=3. Возможно, есть пути синтеза в 2 и 4 стадии, которые пользователь сможет адаптировать под свой запрос, а путей синтеза в 3 стадии может не найтись.


В результате расчета выводится 5 схем с известными реакциями для получения введенного пользователем продукта. Для просмотра полученных данных выберите вкладку со схемой.
На странице отобразятся пронумерованные структуры веществ-реагентов и вещества, полученного в ходе реакции для выбранной стадии.
В таблице для каждого вещества указывается название, его расчетная стоимость в долларах, требуемое количество вещества для реакции, возможный поставщик и ссылка на подтверждение цены.
Внизу таблицы предсказывается общая стоимость продукта.
На странице отобразятся пронумерованные структуры веществ-реагентов и вещества, полученного в ходе реакции для выбранной стадии.
В таблице для каждого вещества указывается название, его расчетная стоимость в долларах, требуемое количество вещества для реакции, возможный поставщик и ссылка на подтверждение цены.
Внизу таблицы предсказывается общая стоимость продукта.
Подсказки:
- Пользователь может редактировать любые данные в таблице (в том числе добавить стадию синтеза), нажав на кнопку «Редактировать таблицу». С момента внесения правок в таблицу она считается отредактированной, она не сохраняется в личном кабинете и ее необходимо скачать, чтобы сохранить данные.
- При отсутствии поставщика система рассчитывает примерную стоимость при помощи алгоритмов искусственного интеллекта. Результаты выбранной схемы можно скачать в файлы форматов .pdf, .csv, .xlsx, нажав кнопку «Скачать» внизу таблицы.
Модуль PDF2SMILES 2.0: Автоматическое извлечение структурных формул из документов в формате PDF
В данном разделе содержится инструмент автоматического распознавания структурных формул молекул в документах в формате .pdf: патентах, научных статьях, протоколах испытаний, диссертациях и т.п.
Модуль обеспечивает оптическое распознавание структур химических соединений и структур Маркуша.
Модуль содержит 2 вкладки “Общие” и “Личные”.
Во вкладке “Общие” содержатся примеры распознавания молекул, которые предоставлены командой Синтелли, во вкладке "Личные" находятся документы, добавленные пользователем.
Модуль обеспечивает оптическое распознавание структур химических соединений и структур Маркуша.
Модуль содержит 2 вкладки “Общие” и “Личные”.
Во вкладке “Общие” содержатся примеры распознавания молекул, которые предоставлены командой Синтелли, во вкладке "Личные" находятся документы, добавленные пользователем.
В правой части расположены распознанные структуры. Их можно выделить и скачать или сохранить в датасет для последующей работы с ними.
Подсказка:
Обратите внимание!
Распознанную системой молекулу можно открыть в молекулярном редакторе и внести правки в структур, при наличии неточностей распознавания, нажав
Подсказка:
- Чтобы выделить 1 или несколько молекул зажмите ctrl или shift и кликните левой кнопки мыши по молекуле.
Обратите внимание!
Распознанную системой молекулу можно открыть в молекулярном редакторе и внести правки в структур, при наличии неточностей распознавания, нажав

Чтобы загрузить свой документ на распознавание структур, необходимо нажать кнопку

После загрузки документа система запускает процесс распознавания структур в нем, который (в зависимости от количества страниц и структур) может занять время от 1 до 8 минут.
Распознанный документ выглядит следующим образом:
Распознанный документ выглядит следующим образом:


11 Раздел «SMILES в IUPAC»
В данном разделе можно автоматически перевести строку SMILES в наименование IUPAC.
В меню слева выберите «SMILES в IUPAC».
В меню слева выберите «SMILES в IUPAC».
В поле ввода напечатайте строку SMILES и нажмите кнопку «Конвертировать».
В качестве результата Система предлагает 5 вариантов IUPAC наименования.
Для каждого варианта названия указан коэффициент точности его предсказания:
В качестве результата Система предлагает 5 вариантов IUPAC наименования.
Для каждого варианта названия указан коэффициент точности его предсказания:


12 Раздел «Статистика»
В разделе собраны статистические параметры моделей, используемых в системе.
Мы придерживаемся открытой политики в части метрик наших моделей. Статистические параметры моделей представлены отдельным модулем на платформе в разделе «Статистика». Мы используем метрики: RMSE, ROC AUC.
RMSE – метрика среднеквадратичной ошибки (чем она меньше, тем лучше). Разберем на примере парацетамола, модель Mouse Intraperitoneal LD50 прогнозирует значение показателя 472 мг/кг. Если наша метрика RMSE по модели Mouse Intraperitoneal LD50 составляет 0.486 это означает, что значение показателя может отклоняться на погрешность в пол порядка. Как считаем: 10 возводим в степень (0,486) и умножаем/делим показатель (472) – получаем погрешность от 150 до 1500 мг/кг. Такая оценка позволяет заведомо отфильтровать высокотоксичные соединения, значения которых обычно лежат ниже 50 мг/кг.
ROC AUC – если ROC AUC показывает 0,5 – то это случайный прогноз (50/50). Чем ближе значение к единице, тем точнее прогноз. Если ROC AUC равен 1 – это идеальная модель. Этот показатель информативнее, чем точность, потому что он учитывает не только результат самого прогноза но еще и уверенность модели в прогнозе.
Для просмотра статистики, в меню слева выберите раздел «Статистика».
Для отображения нужных параметров нажмите на соответствующий раздел.
Мы придерживаемся открытой политики в части метрик наших моделей. Статистические параметры моделей представлены отдельным модулем на платформе в разделе «Статистика». Мы используем метрики: RMSE, ROC AUC.
RMSE – метрика среднеквадратичной ошибки (чем она меньше, тем лучше). Разберем на примере парацетамола, модель Mouse Intraperitoneal LD50 прогнозирует значение показателя 472 мг/кг. Если наша метрика RMSE по модели Mouse Intraperitoneal LD50 составляет 0.486 это означает, что значение показателя может отклоняться на погрешность в пол порядка. Как считаем: 10 возводим в степень (0,486) и умножаем/делим показатель (472) – получаем погрешность от 150 до 1500 мг/кг. Такая оценка позволяет заведомо отфильтровать высокотоксичные соединения, значения которых обычно лежат ниже 50 мг/кг.
ROC AUC – если ROC AUC показывает 0,5 – то это случайный прогноз (50/50). Чем ближе значение к единице, тем точнее прогноз. Если ROC AUC равен 1 – это идеальная модель. Этот показатель информативнее, чем точность, потому что он учитывает не только результат самого прогноза но еще и уверенность модели в прогнозе.
Для просмотра статистики, в меню слева выберите раздел «Статистика».
Для отображения нужных параметров нажмите на соответствующий раздел.

13 Выбор языка
Вы можете настроить язык интерфейса Системы (русский или английский). Для этого в меню слева нажмите на язык и в раскрывающемся меню выберите нужный вариант.

14 Нижнее меню управления профилем
В нижнем меню управления Пользователя можно сменить пароль или выйти из Системы, отредактировать личные данные, а также получить интересующую информацию о Системе. Чтобы открыть это меню, нажмите кнопку
рядом с именем пользователя и в раскрывающемся меню выберите нужный пункт:
рядом с именем пользователя и в раскрывающемся меню выберите нужный пункт:


● «О нас» — осуществляется переход по ссылке на сайт https://syntelly.ru/;
● «Мы в Telegram» — осуществляется переход в телеграм-канал https://t.me/syntelly, где можно задать вопрос разработчикам системы;
● «Руководство пользователя» — открывается актуальная инструкция по пользованию системой;
● «Профиль» — переход на личную страницу пользователя, где можно изменить имя, должность, организацию, а также сменить пароль для входа в систему или выйти из системы.
На этом пока все. Надеемся, что это было полезно!) Если у вас остались вопросы, пишите нам в тг-канал или на почту admin@syntelly.com
Обещаем помочь!
● «Мы в Telegram» — осуществляется переход в телеграм-канал https://t.me/syntelly, где можно задать вопрос разработчикам системы;
● «Руководство пользователя» — открывается актуальная инструкция по пользованию системой;
● «Профиль» — переход на личную страницу пользователя, где можно изменить имя, должность, организацию, а также сменить пароль для входа в систему или выйти из системы.
На этом пока все. Надеемся, что это было полезно!) Если у вас остались вопросы, пишите нам в тг-канал или на почту admin@syntelly.com
Обещаем помочь!